Téměř veškerý chemický průmysl je postaven na katalytických procesech. Rozlišujeme katalýzu homogenní, kdy je katalyzátor rozpuštěn v kapalné fázi – příkladem je výroba valné většiny polymerů – a katalýzu heterogenní, kdy je katalyzátor ve formě tablet, přes které přechází proud reaktantů nebo je případně ve formě prášku, rozptýleného v kapalné nebo plynné fázi. Tento text se zaměří na heterogenní katalytickou hydrogenaci.
Zatímco průmyslové procesy jsou vesměs dokonale zvládnuty, o vlastním průběhu katalytického působení se toho mnoho neví. Jeden z mých profesorů kdysi prohlásil: „Ještě v době nedávné každý, kdo měl díru v zadnici, vymýšlel svou teorii katalýzy. Zaplať Pán Bůh, že už to skončilo“. Neměl tak docela pravdu. Chemická literatura nezadržitelně roste, každým dnem jsme zavalování novými poznatky. Je třeba hledat prostředky, jak tyto poznatky klasifikovat a třídit. Hledání jakéhosi Systema Naturæ (dle terminologie Carl von Linného, 1735) je tedy úsilí oprávněné.
Podívejme se tedy na nejběžnější představy o mechanismu heterogenní hydrogenace. Katalytický povrch tvoří zpravidla plocha kovových atomů v trojúhelníkovém sponu. Teorie multipletů tvrdí, že centrem katalytické aktivity nejsou jednotlivé kovové atomy, nýbrž celá uskupení atomů:
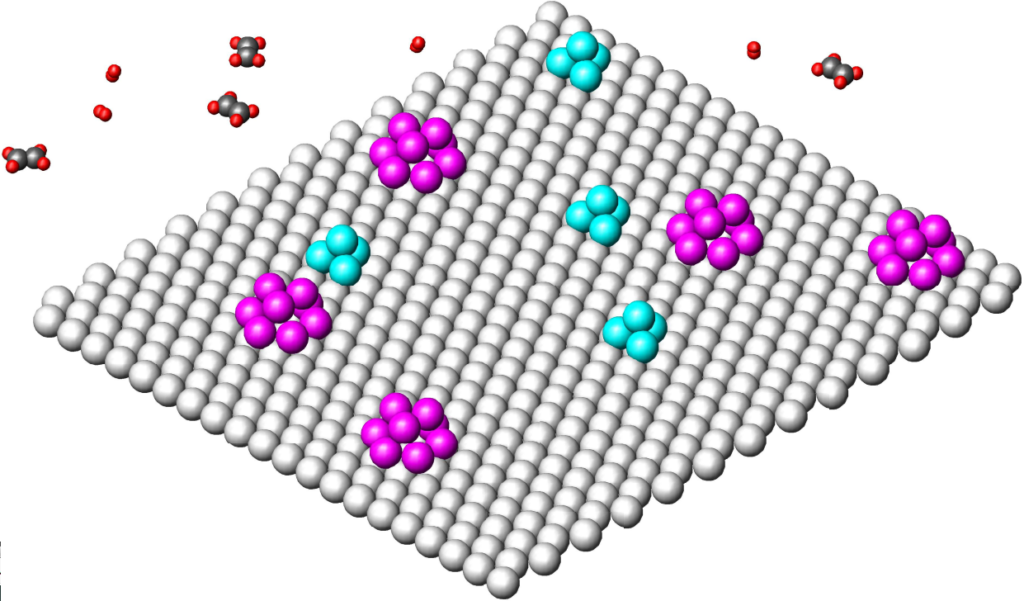
Z důvodů daných kinetickými zjištěními (viz dále: nultý řád k oběma komponentám) se soudí, že na povrchu není jen jeden typ multipletů, nýbrž nejméně dva druhy multipletů, z nichž jeden je schopen pouze aktivovat vodík a druhý pouze olefin; to je tak zvaná „teorie dvou povrchů“. Náš obrázek představuje takový povrch s multiplety dvojí barvy, dvojího druhu. (Poznámka: Molekuly etylénu a vodíku jsou zakresleny s těmito meziatomové vzdálenosti [Å tj.10-10m]: H-H 0,56; C=C 1,33; C-C 1,54; C-H 1,05; v kovové mřížce volíme kov-kov 2,6.) Řekněme, že fialové multiplety jsou schopny aktivovat pouze olefin a ty světle modré pouze vodík. Aktivované molekuly je pak potřeba dostat do vzájemného kontaktu, což je od pohledu nemožné. Leda by měl pravdu onen profesor z Montpellier (Pierre Brun: Catalyse et catalyseurs en chimie organique, Masson 1970), který soudil, že reakční složky pouhým přiblížením k povrchu získají čarovnou sílu, která jim pak umožňuje reagovat v prostoru, mimo katalytický povrch.
Prakticky v každé učebnici organické chemie nalezneme tak zvanou „teorii polohydrogenovaného stavu“ (Horiuti a Polanyi, 1934). Takto ji představuje Wikipedia.org pod heslem „Hydrogenation“:
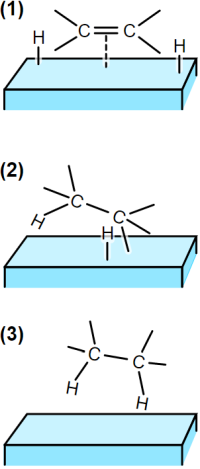
Legenda:
- Reaktanty jsou adsorbovány na povrchu katalyzátoru a H2 (1)
- Jeden atom H se váže na atom C, druhý C zůstává vázán na povrch (2)
- Druhý atom C se váže na druhý atom H. Molekula opouští povrch (3)
Teorie polohydrogenovaného stavu má vysvětlit, proč během hydrogenace olefinů často dochází k izomerizaci: při vratné reakci z (2) na (1) má podle této teorie docházet k odštěpení jiného vodíku než toho, který byl předtím navázán.
Ještě jedno zobrazení téže teorie:
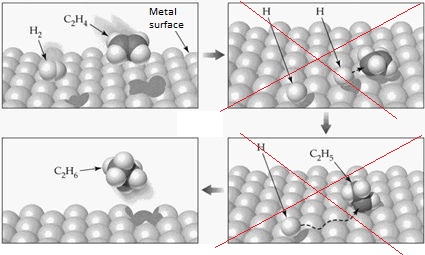
Mám nejméně sedm důvodů se domnívat, že tato teorie je chybná.
První výhrada (důvod č.1):
stejná jako u teorie dvou povrchů – mohou vůbec fragmenty molekul putovat po povrchu tak, jak tento mechanismus vyžaduje?
Princip mikroskopické reversibility říká, že běží-li reakce jedním směrem přes určité meziprodukty, pak reakce ve směru opačném poběží přes stejné meziprodukty. Jistě, v případě hydrogenace je pro vyvolání opačné reakce nutno hodně zvýšit teplotu, ale proč by měl vodík po odštěpení putovat po povrchu kamsi daleko?
Druhá výhrada (důvod č.2):
bylo pozorováno, že při hydrogenaci 1-olefinů v kapalné fázi lze zvyšováním tlaku vodíku dospět do stavu, kdy je reakce nultého řádu vzhledem k oběma komponentám, tedy jak vůči koncentraci olefinu, tak vůči tlaku vodíku. Jak ukážeme níže, při konkurenční adsorpci obou komponent na povrchu je to vyloučeno.
Třetí výhrada:
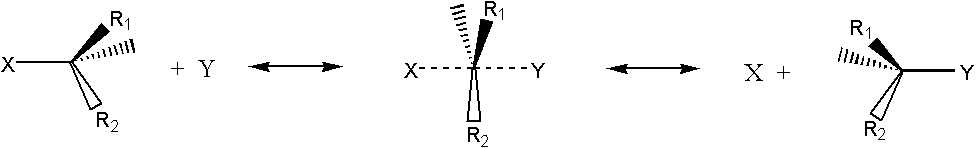
Organičtí chemici se učí, že mezi výchozím a konečným stavem chemické reakce je jenom jeden přechodový stav a tento je co možná symetrický (důvod č.3). Jako příklad uvedeme Waldenovu reakci, tedy výměna halogenu X za halogen Y. Pokud jsou substituenty na uhlíku rozdílné, vykazuje molekula optickou otáčivost, přičemž reakcí dochází k její inverzi, což prokazuje přístup halogenu Y z jedné strany a odštěpení halogenu X ze strany opačné:
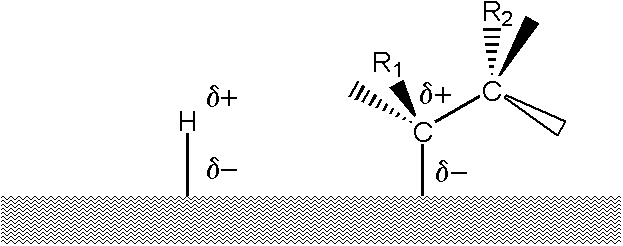
My se domníváme, že analogický mechanismus je dobře možný i v případě hydrogenace:
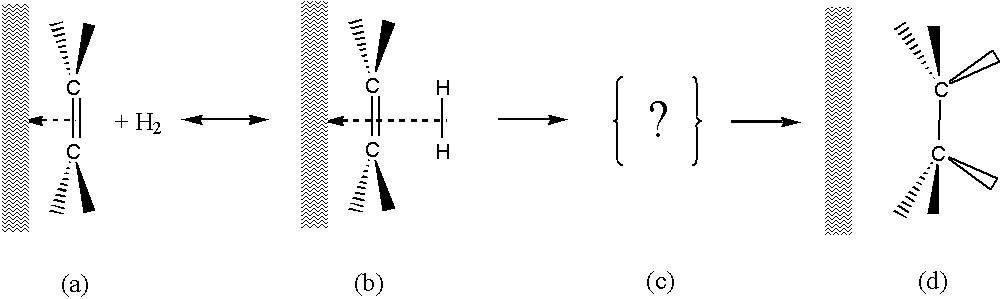
- Olefin je adsorbován na kovový povrch – silná chemická vazba. (a)
- Vodík je adsorbován ve druhé vrstvě – fyzikální až chemická vazba. (b)
- Přechodový stav, kdy oslabuje vazba olefinu s povrchem a současně se zesiluje vazba s vodíkovými atomy. Substituenty na uhlících se přitom vychylují směrem ke katalytickému povrchu, což může předčasně oslabit vazbu olefinu na povrch. Proto tedy hydrogenace 1-olefinů a dvojné vazby se dvěma substituenty probíhá, zatímco hydrogenace tri– a tetra- substituovaných dvojných vazeb je vyloučena. (c)
- Nasycená molekula opouští povrch. (d)
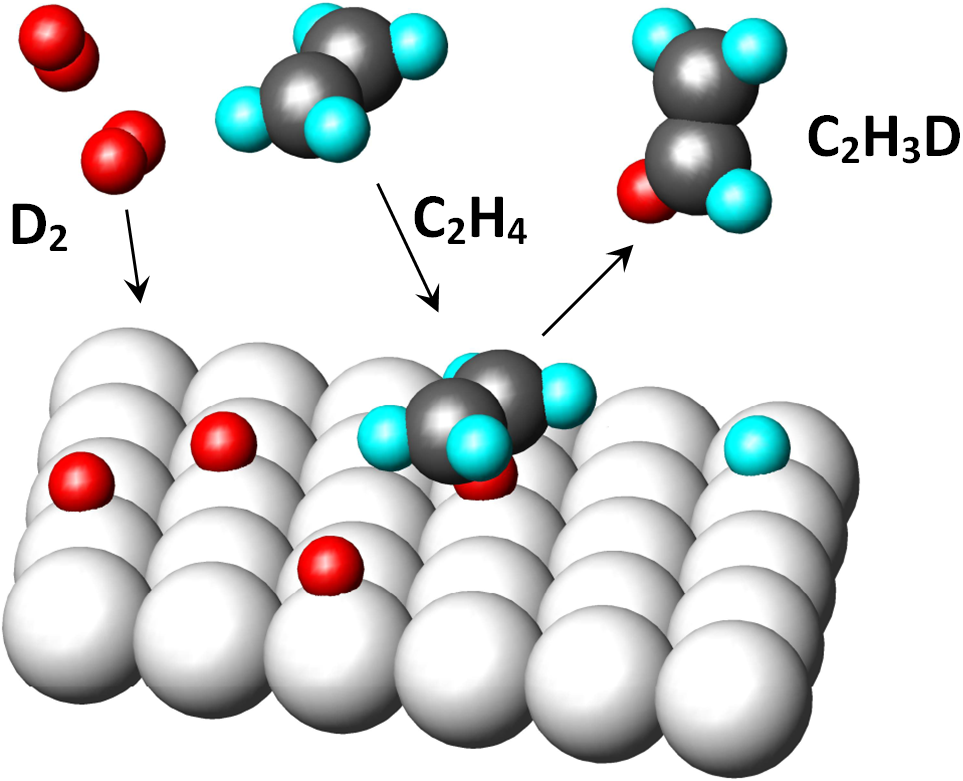
Kalotový model:
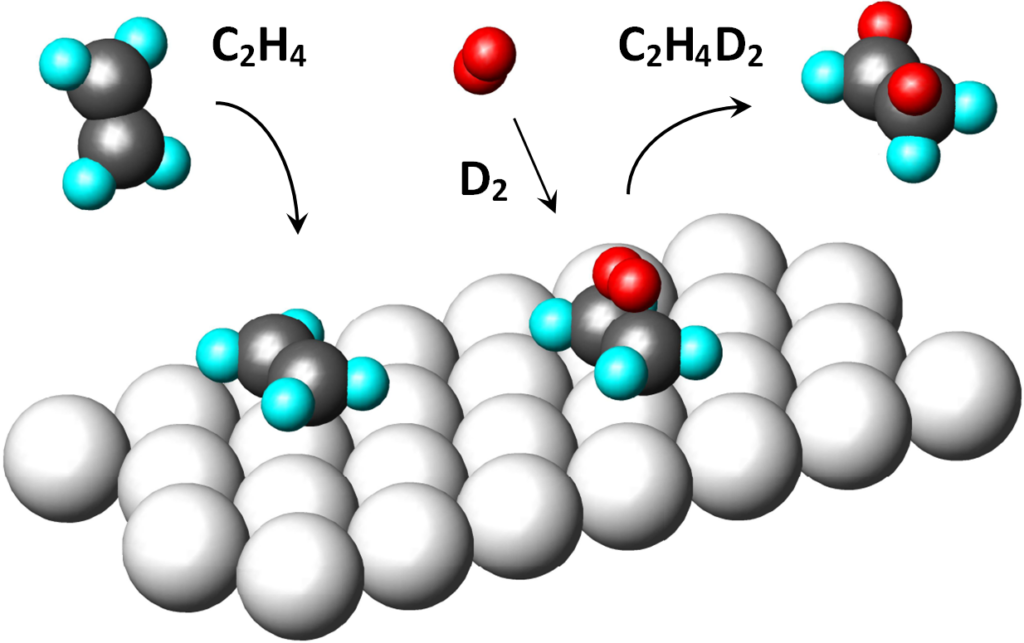
Na obrázku jsou původní vodíky světle modře, nově navázané vodíky (D2) červeně. Jedná se o cis-adici celistvé molekuly vodíku.
Bylo pozorováno, že kupř. na platinovém katalyzátoru při hydrogenaci směsí molekul H2 + D2 vzniká produkt, který obsahuje buďto žádný, nebo dva atomy deuteria. Toto pozorování je jen obtížně slučitelné s teorií polohydrogenovaného stavu (důvod č.4).
Ještě se krátce vrátíme k předchozí teorii, k teorii multipletů. Ta byla založena na pozorování konverze benzen/cyklohexan. Tato reakce je katalyzována pouze kovy, jejichž povrch tvoří trojúhelníkovou strukturu se vzdáleností mezi sousedními atomy mezi 2,40 až 2,77 Å. Kovy se vzdálenostmi mimo toto rozmezí nebo s jinou geometrií povrchu jsou neaktivní. Do takovéto struktury lze pak pěkně zakreslit šestiúhelník, který představuje šestici uhlíků:
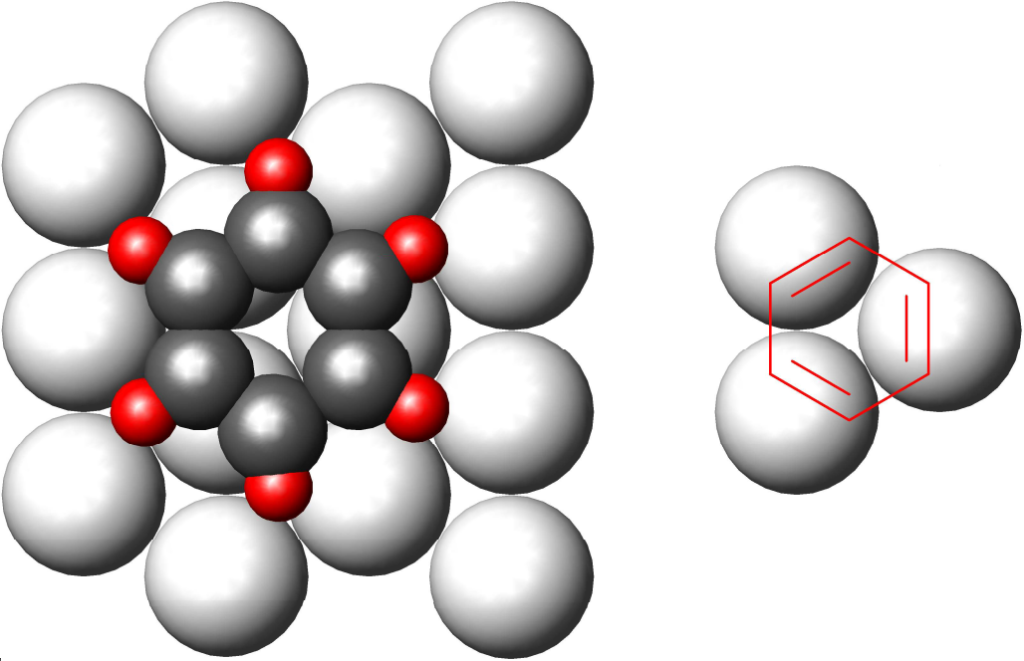
Leč: pokud na benzenové jádro pohlížíme jako na seskupení tří dvojných vazeb, pak se jen potvrzuje pravidlo, že na hydrogenaci jedné dvojné vazby postačí jen jeden atom kovu.
Nyní se budeme věnovat izomerizaci. Izomerizací může být reakce izotopové výměny, cis/trans izomerizace či posun dvojné vazby v uhlíkovém řetězci. V homogenním prostředí je izomerizace vratnou reakcí katalyzovanou vodíkovým kationtem:
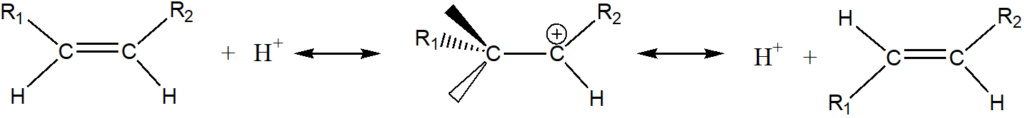
V případě heterogenní reakce na kovových površích v přítomnosti vodíku musíme předpokládat, že izomerizace je reakcí nezávislou na hydrogenaci, katalyzovanou – stejně jako v homogenním prostředí – protonickým vodíkem. Přítomnost protonického vodíku na povrchu katalyzátoru je přinejmenším vysoce pravděpodobná. Vzpomeňme třeba skutečnost, že vodík rozpuštěný v kovu putuje vlivem elektrického proudu, což je pokládáno za důkaz jeho přítomnosti v „kovové“, tedy protonické formě. Izomerizační reakce mohou pak probíhat s meziproduktem typu
Proč se v tomto případě nejedná o polohydrogenovaný stav? Jde zde o elektrofilní adici, kdy dvojná vazba C=C představuje nukleofilní činidlo. Příklady redukce dvojné vazby redukčními činidly, tedy iontové hydrogenace, existují. V katalytické hydrogenaci by to však vyžadovalo heterolytické štěpení molekuly vodíku (H+, H–), což je na povrchu tvořeném atomy jediného kovu celkem nemyslitelné – připomeňme si v této souvislosti termíny vodíková elektroda, Nernstova rovnice. Takže ve chvíli, kdy přisoudíme atomárnímu vodíku vázanému na povrch katalyzátoru kladný náboj, teorie polohydrogenovaného stavu padá (důvod č.5). Pozor: toto platí pro homogenní povrchy tvořené jediným kovem, v případě povrchu modifikovaným jiným kovem to platit nemusí, jak zjistil již před sto lety Adams s katalyzátorem platina modifikovaná železem.
Navíc, v případě jednoduchých olefinů (především na Pt) je hydrogenace v kapalné fázi syn-adicí, vzniká tedy produkt cis-, zatímco izomerace meziproduktu, jako k ní dochází během iontové hydrogenace, vede k produktu trans- (důvod č.6).
Kalotový model izomerizace:
Celkové množství protonického vodíku na povrchu katalyzátoru patrně souvisí se schopností jednotlivých kovů vodík rozpouštět. Extrémní je palladium, které je schopno rozpouštět 350 – 850 krát více vodíku, než činí jeho vlastní objem; tuhý roztok vodíku v palladiu o složení Pd2H je dokonce pokládán za definovanou sloučeninu. Naproti tomu v platině se rozpouští za normálních teplot a tlaku řádově jen 10-7cm3 H2/cm3 Pt. Je tu patrná souvislost se skutečností, že na platině probíhají izomerizační reakce (a také hydrolytické reakce) jen v malé míře, na paladiu zpravidla ve značné míře. Při hydrogenaci na úkor vodíku rozpuštěného v katalyzátoru bylo zjištěno, že takto vzniká vždy více izomerů (migrace dvojné vazby) než při běžné hydrogenaci pod vodíkem na témže katalyzátoru. Podle jedné studie se na černích osmiové, iridiové, platinové, rutheniové a rhodiové (čerň – oxid kovu, redukovaný in situ) zúčastňuje při přemisťování dvojné vazby především silně adsorbovaná forma vodíku, tedy vodík atomární, zatímco v hydrogenaci slabě adsorbovaná forma, tedy vodík molekulární (důvod č.7).
Bylo zjištěno, že pokud je kov před hydrogenací olefinu zbaven vodíku, ztrácí izomerizační schopnosti. Niklový katalyzátor, na němž byl předtím hydrogenován 1-oktin ztratí schopnost izomerizace 1-hexenu, zatímco jeho schopnost hydrogenovat tento olefin zůstává nezměněna (důvod č.8).
Izomerizace je rovnovážnou reakcí. V cis/trans izomerizaci se uplatňuje především sterický efekt, rovnováha proto směřuje výrazně ke struktuře trans-. Směr migrace dvojné vazby je zase dán polarizací dvojné vazby působením indukčního efektu – v případě přesunu vodíku na jiné místo v uhlíkovém řetězci platí ono biblické „Kdo má málo, tomu bude vzato“ (bible, Matouš 25:29). Při interpretaci experimentálních dat je ale třeba si uvědomit, že izomerizace je rovnovážná reakce, dochází tedy i k izomerizaci dvojné vazby z místa větvení na rovný úsek. Izomerní olefin se pak může stabilizovat přímou vazbou na katalytický povrch a být následně hydrogenován. Tri-substituované olefinické sloučeniny, které se nedaří hydrogenovat na platině, mohou být proto hydrogenovány na paladiu, případně na kovu naneseném na kyselém nosiči alumina (γ-Al2O3) nebo ve směsi s aluminou.
Kinetický model podle Langmuira a Hinshelwooda
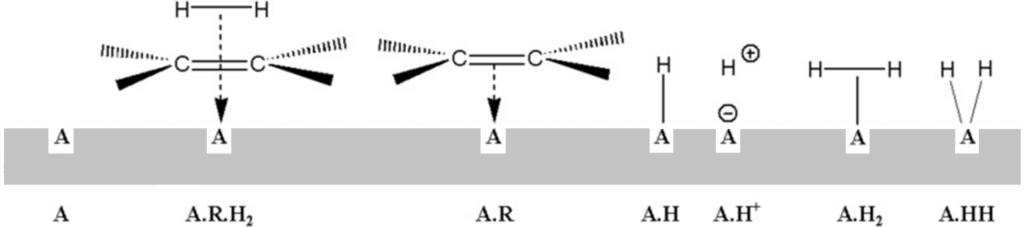
V rychlostních rovnicích homogenních reakcí vystupují v roli aktivit zpravidla koncentrace. Pro reakce na površích definuje Hinshelwood aktivitu jako podíl povrchových aktivních míst, obsazených příslušným reaktantem, vůči celkovému počtu aktivních míst povrchu. Adsorpce na povrchu je konkurenční a probíhá podle Langmuirovy adsorpční izotermy. Předpokládejme kupříkladu, že se na povrchu nacházejí tyto adsorpční komplexy:
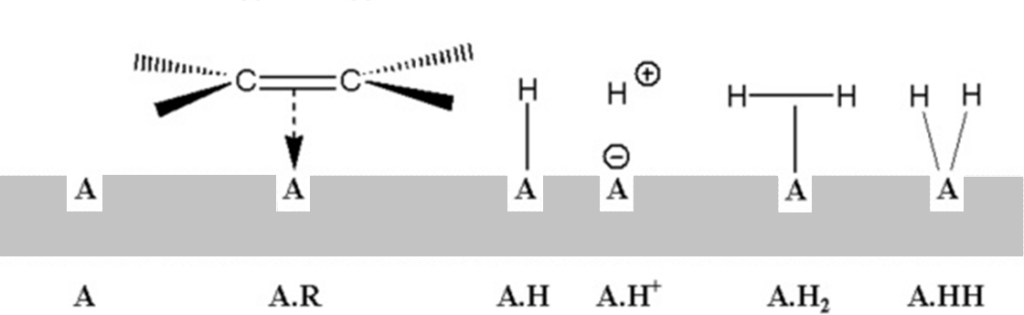
Langmuirovská bilance, vyjádřená nejprve graficky a pak rovnicí:

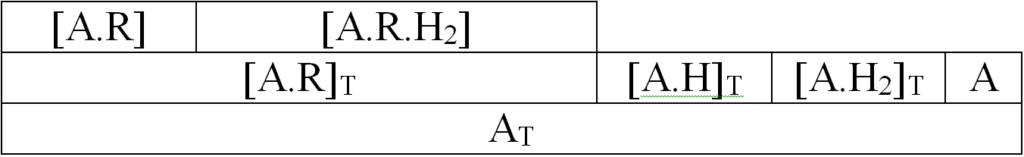
[A]T = [A] + [A.R]T + [A.H]T + [A.H2]T [1.1]
kde:
[A]T celkový počet aktivních míst povrchu
[A] aktivní místa neobsazená
[A.R]T aktivní místa obsazená olefinem
[A.H]T aktivní místa obsazená atomárním vodíkem (A.H, A.H+)
[A.H2]T aktivní místa obsazená molekulárním vodíkem (A.H2, A.HH)
Pro každou adsorbovanou složku lze definovat konstantu adsorpční rovnováhy:
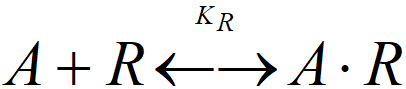
KR = [A.R]T/([A]·c)
kde c je aktivita (koncentrace) olefinu v roztoku
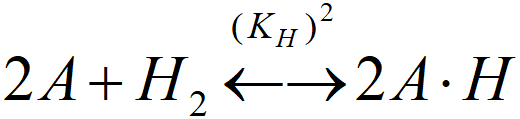
(KH)2 = [A.H]T2· /([A]2·PS)
Kde PS je aktivita vodíku (jeho parciální tlak u povrchu katalyzátoru, viz další kapitola)
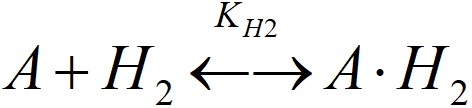
KH2 = [A.H2]T/([A]·PS)
Dosazením do výchozí bilanční rovnice dostáváme
[A]T = [A] + [A]·KR·c + [A]·KH·PS0.5 + [A]·KH2·PS
Aktivita olefinu na katalytickém povrchu pak činí
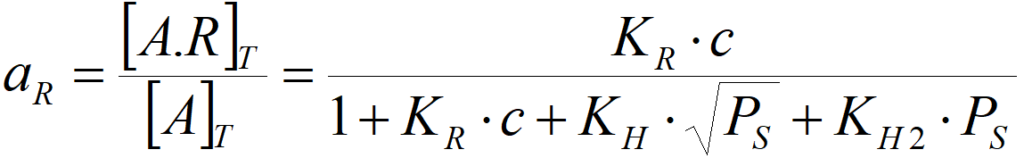
a podobně pro atomární vodík
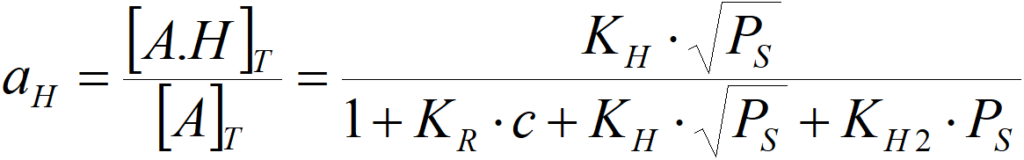
Pokud jsou prakticky všechna aktivní centra povrchu pokryta olefinem,
tj. KR·c >> 1+KH·PS0,5+KH2·PS,
pak aR=1 a další zvyšování koncentrace již aktivitu prakticky nemění, tj. reakce je nultého řádu k olefinu. Aktivita vůči vodíku je pak (pro atomární vodík) aH = KH·PS0,5/(KR·c) (přičemž podíl 1/KR·c je v tomto případě prakticky konstantní) neboli řád reakce vůči vodíku bude nenulový. Obdobně, pokud je povrch pokryt vodíkem a deficitní je olefin, pak řád reakce vůči vodíku bude nulový, avšak řád vůči olefinu nemůže být nulový. Platí to zcela obecně: při konkurenční adsorpci dvou reaktantů nemůže být řád reakce nulový zároveň vůči oběma dvěma.
Pokud byl tedy při hydrogenaci olefinů za vyššího tlaku pozorován nultý řád reakce vůči oběma reaktantům pak jak jsme již dříve uvedli, toto není slučitelné s teorií polohydrogenovaného stavu.
Kinetický model dvojvrstevné adsorpce
V rychlostních rovnicích podle Langmuira a Hinshelwooda jsme mlčky přijali různá zjednodušení. Tak počet aktivních míst povrchu bereme za konstantní, nezávislý na stupni obsazení povrchu jednotlivými adsorbovanými látkami a všechna aktivní centra pokládáme za rovnocenná. Tato a další zjednodušení však nemají vliv na rámcovou (tedy kvalitativní, nikoli kvantitativní) platnost našich závěrů.
Rozšíříme nyní Hinshelwoodovu teorii na dvouvrstevnou adsorpci. Uvažujeme následující povrchové komplexy:
Grafické zobrazení bilance obou vrstev:
Bilanci první vrstvy jsme již řešili. Předpokládáme, že adsorpcí vodíku v druhé vrstvě se nemění rovnováha ve vrstvě první. Bilance druhé (horní) vrstvy:
[A.R]T = [A.R] + [A.R.H2]
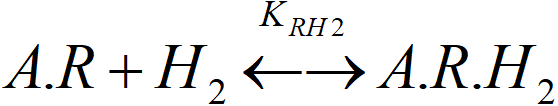
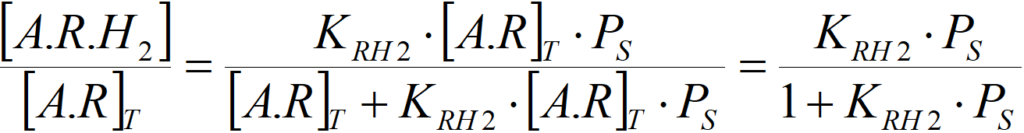
Vynásobením levých a pravých stran této rovnice s výše uvedenou rovnicí pro aR = [A.R]T/[A]T dostáváme
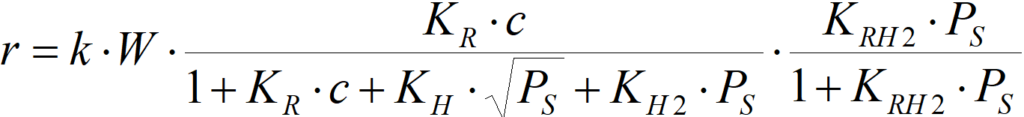
W je množství katalyzátoru
k je rychlostní konstanta; zahrnuje i množství aktivních center AT, připadajících na jednotkové množství katalyzátoru W.
Rovnici je nutno chápat jako semikvantitativní – při jejím odvození bylo přijato hodně zjednodušení. Platí nicméně, že
pokud KR·c >> 1+KH·PS0,5+KH2·PS a KRH2·PS >> 1, pak
r = k·W [1.6]
čili: nultý řád k oběma reagujícím složkám, v jistém rozsahu koncentrací reagujících složek, je možný.
Uzavřeme tedy, že i z kinetického hlediska naše teorie dvouvrstevného aktivovaného komplexu vyhovuje. Naproti tomu k všeobecně přijímané „teorii polohydrogenovaného stavu“ doporučujeme přistupovat (slušně řečeno) jen s velkou rezervou .
Jiné funkční skupiny
Nežli opustíme teoretickou část věnovanou katalytickému působení, vrátíme se k úloze vodíku vázaného přímo na katalytický povrch. Jestliže námi navržený model katalytické hydrogenace olefinických sloučenin vylučuje přímou účast atomického vodíku, což se týká i vodíku rozpuštěného v mase kovu, pak je třeba zdůraznit, že pro jiné funkční skupiny, často silně polarizované, tomu bude jinak. Příkladem takové hydrogenace, kterou si lze těžko představit bez účasti protonického vodíku, je hydrogenace směsi benzaldehyd/dimetylamin, která dá dimetylbenzylamin. Na platině dává reakce výtěžek kolem 90%, ale na rhodiu nebo palladiu je selektivita nad 99%:
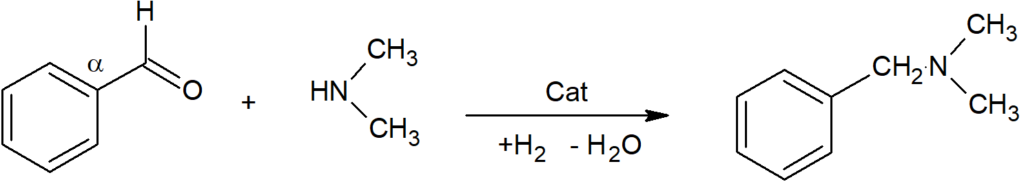
Zdá se, že zde před odštěpením vody – vytvořením Shiffovy báze, která je pak dále hydrogenována, dochází k vytvoření amoniového komplexu povrchového atomárního vodíku s aminem, čili protonický vodík je katalyticky účasten přímo v hlavní reakci.
Jak už jsme zmiňovali v jiných souvislostech, při hydrogenaci acetylenických sloučenin, tedy trojné vazby mezi uhlíky, je povrchový vodík přímo spotřebováván. Na platině běží tato reakce velmi pomalu po celou dobu přítomnosti trojné vazby, pak ale rychlost reakce prudce vzroste a hydrogenace dvojné vazby proběhne v krátké době.
Závěrem zopakujme, že zkoumání reakčních mechanismů heterogenní katalýzy je půda značně nejistá. Naproti tomu Hinshelwoodova teorie povrchových aktivit se zdá být spolehlivá.